
Comparative Genomics
New comparative genomic methods
Institution: Senckenberg Gesellschaft für Naturforschung
Prof. Dr. Michael Hiller
Powerful computational methods are an important basis for gaining new insights through comparative genome analyses. In previous years, the Hiller lab has developed several new approaches, for example to detect gene losses in genome alignments, to find associations between phenotypic differences and differences in regulatory elements and to improve sensitivity and specificity of whole genome alignments.
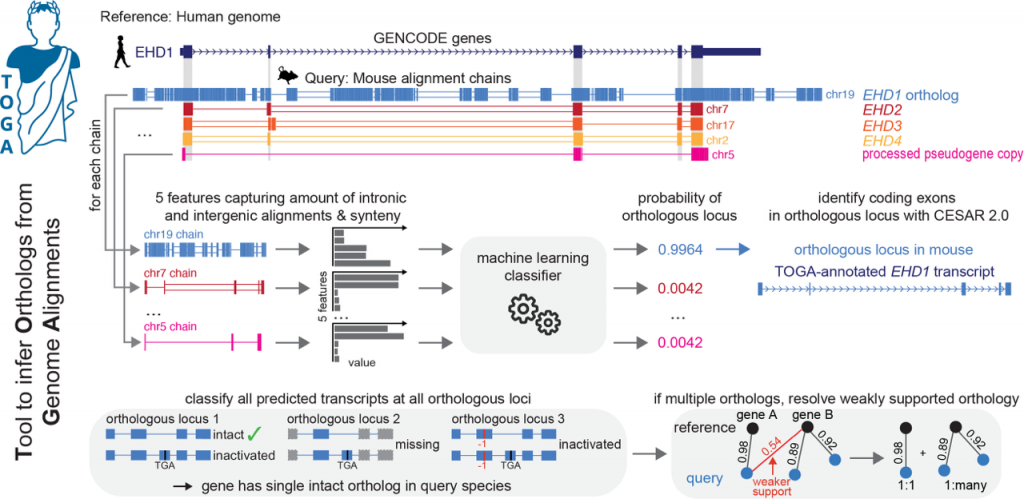
In our current work, we have developed TOGA (Tool to infer Orthologs from Genome Alignments), the first method that integrates gene annotation and ortholog inference. TOGA implements a novel methodology to infer orthologous gene loci between related species that does not rely on protein or coding exonic sequences. Instead TOGA utilizes whole genome alignments and machine learning to accurately distinguish orthologs from paralogs or processed pseudogenes based on alignments of intronic and intergenic regions. TOGA scales to many genomes, which enabled us to generate gene annotations and ortholog sets across more than 500 mammals and 400 birds, creating the largest comparative datasets for these clades so far. Additionally, TOGA automatically detects losses of genes and enables screens for positive selection.
We will expand the repertoire of methods to enable a more comprehensive comparative analysis of genome data through the accurate detection of new “types” of functionally relevant genomic changes. This includes the detection of relevant changes in non-coding RNAs as well as the detection of gene duplication events for which we plan to extend our HMM-based CESAR method and its integration into TOGA.
Illustration of the TOGA method
TBG Project team
- Alexis-Walid Ahmed
- Bogdan Kirilenko
- Michael Hiller (PI)
Source code
More information is here.
Publications:
Kirilenko BM, Munegowda C, Osipova E, Jebb D, Sharma V, Blumer M, Morales A, Ahmed AW, Kontopoulos DG, Hilgers L, Zoonomia Consortium, Hiller M. TOGA integrates gene annotation with orthology inference at scale, submitted
A Ludwig A, Pippel M, Myers G, Hiller M. DENTIST – using long reads to close assembly gaps at high accuracy. GigaScience, 11, giab100, 2022