
Home
COMPUTATIONAL BIOLOGY AND EVOLUTIONARY GENOMICS: DISCOVERING PHENOTYPE-GENOTYPE ASSOCIATIONS
Evolution has led to an incredible diversity of phenotypes in all major species clades, exemplified by mammals like bats that fly or dolphins that live their entire life in the water. This phenotypic diversity is the outcome of changes or loss of ancestral phenotypes or gain of novel phenotypes during evolution. Phenotypic differences between species are due to differences in their DNA (genome). Today, the genome of hundreds of animals have been sequenced, including more than 100 mammals and 300 birds, and many more will be sequenced in the near future.
Given that the genome determines many phenotypes of an organism, these sequenced genomes provide an unprecedented opportunity to discover which genomic changes underlie particular phenotypic changes between species. This is the overarching scientific question we address in the lab by combining both computational as well as experimental approaches. Thus, we wish to contribute to our understanding how nature’s incredible diversity evolved. Our focus is here explicitly on differences between species and not differences within a species, though many of our comparative methods will also work on genomes of individuals or strains of the same species.
The complexity and difficulty of this phenotype-genotype question at the between-species level is illustrated by the fact that their genomes differ by millions of changes. Just accurately comparing the genomes of two, not too closely related species poses a big challenge. Despite this, our previous research established a proof-of-concept that makes it possible to detect genomic loci that underlie a particular phenotypic difference. This “Forward Genomics” approach focuses on independently evolved phenotypic differences and uses ancestral sequence reconstruction to systematically search whole genomes for differences that match the given phenotypic patterns.
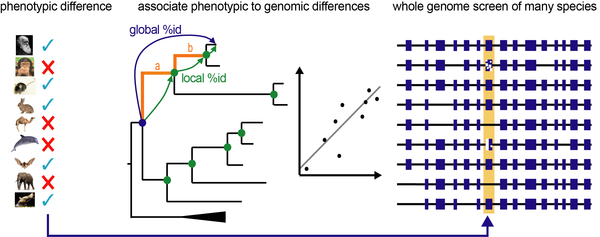
Illustration of our Forward Genomics framework that associates phenotypic differences between species to differences their genomes. We use ancestral sequence reconstruction to measure divergence of functional genomic regions and rely on statistical approaches to detect phenotype-genotype associations.
CURRENT RESEARCH
Current research in the lab combines computational and experimental approaches. On the computational side, we both develop and apply comparative genomics approaches. For example, we are developing several methods to accurately detect those genomic differences that likely change the function of genes or regulatory elements, which can impact species’ biology. Since these methods rely on high-quality whole genome alignments, we developed ways to improve genome alignments by detecting sequence homologies between distant species and filtering out non-orthologous alignments. Our alignment of 144 vertebrate genomes (including 73 non-human mammals, 31 birds and 23 teleost fish) provides a powerful basis to detect both conservation and divergence with comparative genomics. To improve the power of discovering statistical associations between genomic and phenotypic changes in Forward Genomics searches, we developed new methods that control for phylogenetic relatedness between species and evolutionary rate differences. Our computational approaches give us an extensive toolbox for analyzing genomic data, for detecting biologically important differences and for addressing the phenotype-genotype question.
On the experimental side, we use approaches like RNA-seq and ATAC-seq to annotate genes and regulatory regions in specific tissues of interest and use these annotations to interpret our computational results. In addition to making use of the wealth of genomes sequenced by the community, we select species with interesting phenotypes, and sequence and assemble their genomes. Finally, to establish a causal link between a discovered genomic difference and a natural phenotypic difference, we experimentally test if introducing the detected genomic difference in a model organism affects the predicted phenotype.
A main limitation to understand which genomic changes underlie particular phenotypic changes between species is digital access to phenotypic data. To this end, we are currently collaborating with zoologists to address the pressing need to close the phenotype gap for sequenced species.