Tools and Data
TOGA
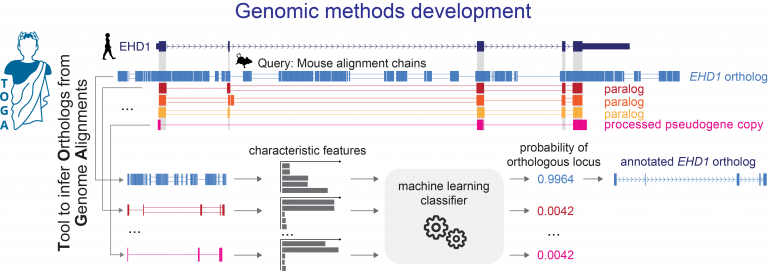
TOGA (Tool to infer Orthologs from Genome Alignments) is the first method that integrates gene annotation, inferring orthologous genes and classifying genes as intact or lost [1]. TOGA implements a novel paradigm to infer orthologous gene loci that largely relies on intronic and intergenic alignments and uses machine learning to accurately distinguish orthologs from paralogs or processed pseudogenes. As a reference-based method, TOGA is scalable and can be applied to hundreds of species. TOGA also detects gene losses and facilitates the generation of more accurate codon alignments to screen for selection patterns. With a set of ancestral genes, TOGA can also provide superior measures of genome quality.
Source code: github
Data for 517 placental mammal and 501 bird assemblies: Comparative gene annotations, ortholog sets, lists of inactivated genes and multiple codon alignments are available for download. Our UCSC browser mirror at https://genome.senckenberg.de/ provides annotation tracks for all analyzed mammal and bird assemblies.
[1] Kirilenko et al. Integrating gene annotation with orthology inference at scale. Science, 380 (6643), 2023
GENE LOSS DETECTION PIPELINE
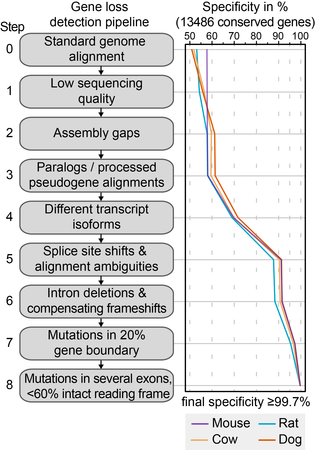
We developed a genomics approach to detect gene-inactivating mutations across many genomes [1]. Accurately detecting such mutations in genome alignments poses a number of challenges. In order to achieve high accuracy, our pipeline implements a number of steps that overcome assembly and alignment issues, and address evolutionary exon-intron structure changes in genes that are conserved. Using a large set of 13,486 human genes that have annotated 1:1 orthologs in mouse, rat, cow and dog, we could show that this approach achieves a specificity of 99.7%.
Source code: github
[1] Sharma V, Hecker N, Roscito JG, Foerster L, Langer BE, and Hiller M. A genomics approach reveals insights into the importance of gene losses for mammalian adaptations. Nature Communications, 9, 1215, 2018
REFORGE
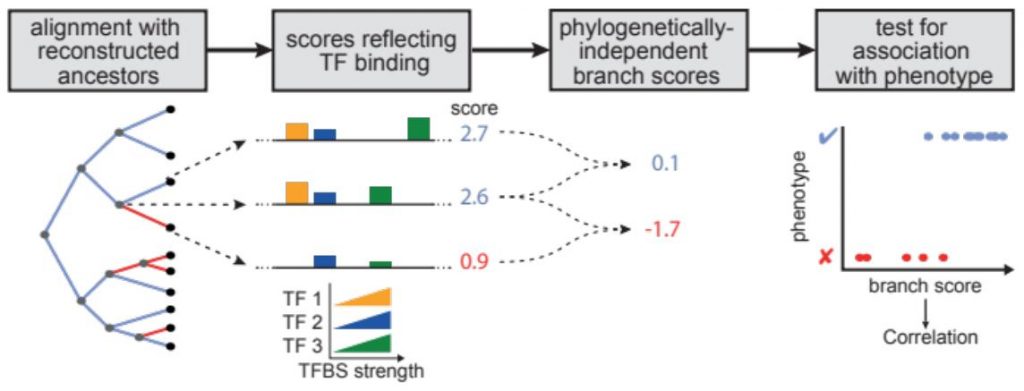
REforge (Regulatory Element forward genomics) is a method to associate transciption factor binding site divergence in regulatory elements with phenotypic differences between species [1].
Like the Forward Genomics branch method [2], REforge uses of ancestral sequence reconstruction to consider evolutionary changes that happened on each individual branch in a phylogenetic tree. In contrast to standard Forward Genomics, REforge does not measure sequence divergence, but estimates differences in the collective binding affinity of a given set of TFs on every branch. This better predicts functional differences in regulatory activity, making REforge the superior method when applied to cis-regulatory elements.
Source code: github
Data (Simulated regulatory element evolution): CBG Big Data Server
[1] Langer BE, Roscito JG, Hiller M: REforge associates transcription factor binding site divergence in regulatory elements with phenotypic differences between species. Mol Biol Evol, 35(12), 3027–3040, 2018
[2] Prudent X, Parra G, Schwede P, Roscito JG, Hiller M. Controlling for phylogenetic relatedness and evolutionary rates improves the discovery of associations between species’ phenotypic and genomic differences. Mol Biol Evol, 33 (8), 2135–2150, 2016
CHAINCLEANER, REPEATFILLER AND A 144-VERTEBRATE ALIGNMENT
Accurate and sensitive alignments between entire genomes are crucial for comparative genomics. First, our chainCleaner method [1] improves the specificity in genome alignments by accurately detecting and removing paralogous and random local alignments that overlap orthologous loci. Applying chainCleaner to alignment chains (i) exposes truly orthologous alignments that have been masked before, (ii) facilitates inferring rearrangements and the evolutionary history of genomes, and (iii) rescues hundreds of kilobases of missing alignments. Second, our RepeatFiller method detects highly-sensitive local alignments improves alignment chains by uncovering remote homologies [2].
Source code: github
We applied chainCleaner and highly-sensitive parameters to build a multiple alignment of 144 vertebrate genomes, including 73 non-human mammals, 31 birds and 23 teleost fish [3]. We also ran CESAR on this 144-vertebrate alignment to get an comparative gene annotation in 143 verbrate genomes at substantially increased sensitivity.
Data (144-vertebrate alignment, human genes mapped to 143 vertebrates): CBG Big Data Server.
Click HERE to load the CESAR comparative gene annotation using the 144-vertebrate alignment into the UCSC genome browser.
FORWARD GENOMICS 2.0
Forward Genomics is our framework to associate phenotypic differences between species to differences in their genomes [1]. We focus on phenotypes that are repeatedly lost or changed in independent lineages. Forward Genomics associates a given phenotypic difference to differences in the genome by searching for genomic regions that likely evolve neutrally in the trait-loss lineages and likely evolve under selection in the trait-preserving lineages. The original Forward Genomics implementation (“perfect match” method) searches for genomic regions where all trait-loss species are more diverged than all trait-preserving species [1].
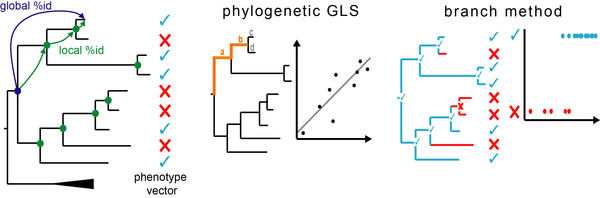
New Forward Genomics methods are:
- “GLS”, which uses a generalized least square approach and
- the “branch method”, which detects phenotype-genotype associations by using per-branch divergence values.
In contrast to perfect-match, GLS and branch method directly control for the phylogenetic relatedness and evolutionary rate differences between species, which substantially improves sensitivity and specificity [2].
Source code: github
Data (Simulated trait losses, Loss of vitamin C and loss of vision data, multiple genome alignment): CBG Big Data Server
[1] Hiller M, Schaar BT, Indjeian VB, Kingsley DM, Hagey LR, and Bejerano G. A “forward genomics” approach links genotype to phenotype using independent phenotypic losses among related species. Cell Reports, 2(4), 817-823, 2012
[2] Prudent X, Parra G, Schwede P, Roscito JG, Hiller M. Controlling for phylogenetic relatedness and evolutionary rates improves the discovery of associations between species’ phenotypic and genomic differences. Mol Biol Evol, 33 (8), 2135–2150, 2016
CODING EXON STRUCTURE AWARE REALIGNER (CESAR) 2.0
CESAR is a tool to realign coding exons with a Hidden-Markov-Model that directly incorporates the reading frame and splice site annotation of each exon [1]. We use CESAR to assess the conservation of coding exons in whole genome alignments, which is a prerequisite to map exon annotations from a reference to numerous aligned genomes. The motivation for developing CESAR was that genome alignments are not aware of the reading frame and the splice sites of exons, which results in numerous spurious frameshift and splice site mutations in exons that are truly conserved in other species. To improve this, CESAR will align the exon again (“realign”), preserving the reading frame and splice sites if the exon is conserved in other species.
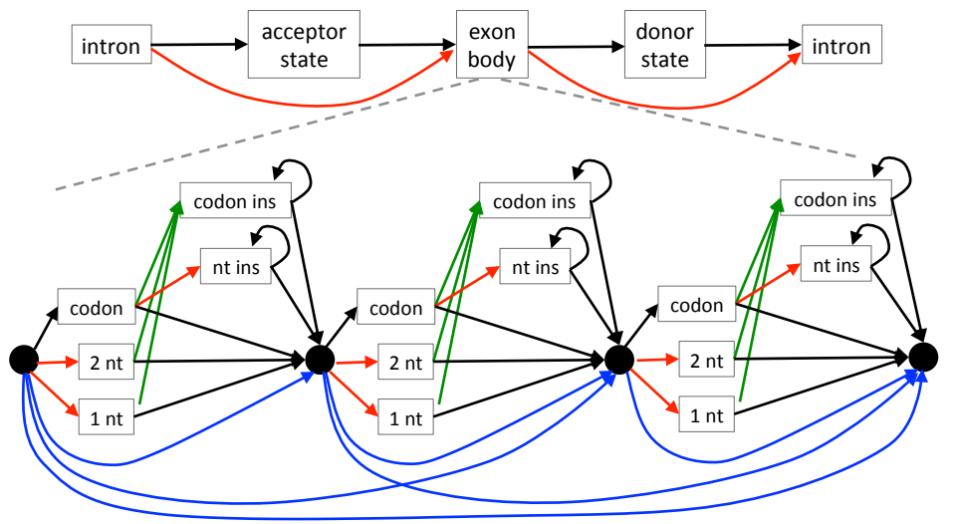
We recently developed CESAR 2.0 [2]. Compared to its predecessor [1], CESAR 2.0 is 77X times faster on average (132X times faster for large exons) and requires 30-times less memory. In addition, CESAR 2.0 improves the accuracy of the comparative gene annotation by two new features. First, CESAR 2.0 substantially improves the identification of splice sites that have shifted over a larger distance, which improves the accuracy of detecting the correct exon boundaries. Second, CESAR 2.0 provides a new gene mode that re-aligns entire genes at once. This mode is able to recognize complete intron deletions and will annotate larger joined exons that arose by intron deletion events.

CESAR 2.0 Source code: github
Data (Test alignments and real splice site shifts from CESAR): CBG Big Data Server
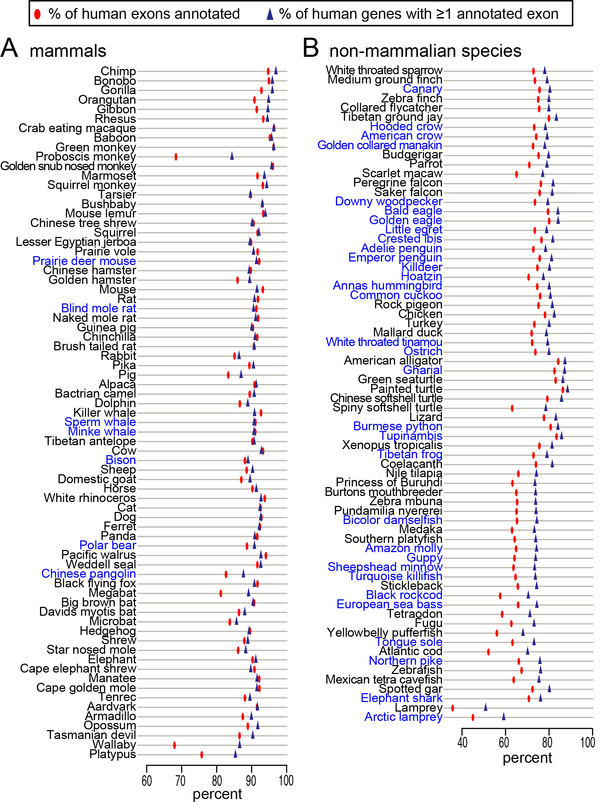
We applied CESAR our 144-vertebrate genome alignment and mapped human exons to 143 non-human vertebrates [3]. This provides an accurate comparative gene annotation for many poorly annotated species and contributes to reduce the growing gap between genome sequencing and genome annotation.
Data (human genes in genePred format mapped to 143 vertebrates): CBG Big Data Server
[1] Sharma V, Elghafari A, Hiller M. Coding Exon-Structure Aware Realigner (CESAR) utilizes genome alignments for accurate comparative gene annotation. Nucleic Acids Res. 44(11), e103, 2016
[2] Sharma V, Schwede P, Hiller M. CESAR 2.0 substantially improves speed and accuracy of comparative gene annotation. Bioinformatics, 33(24), 3985–3987, 2017
[3] Sharma V, Hiller M. Increased alignment sensitivity improves the usage of genome alignments for comparative gene annotation. Nucleic Acids Res, 45(14) 8369-8377, 2017
ITERATIVE CORRECTION OF SEQUENCING ERRORS (SGA-ICE)
A key step in genome assembly is to computationally correct sequencing errors that occur in the sequencing reads. We have developed a new strategy called “iterative error correction” that minimizes the total amount of erroneous reads by simply using multiple correction rounds [1]. This strategy is most effective on long 250 or 300 bp Illumina reads that the MiSeq or new HiSeq technology generates. The higher read accuracy obtained by iterative correction substantially improves contig assembly of long Illumina reads. The SGA-ICE.py tool automates iterative error correction with as little user input as possible. SGA-ICE.py runs multiple rounds of k-mer-based correction with an increasing k-mer size, followed by a final round of overlap-based correction. By combining the advantages of small and large k-mers, SGA-ICE.py can correct more base substitution errors, especially in repeats. The final overlap-based correction round can also correct small insertions and deletions.
Source code: github
Data (error profiles and simulated 300 bp reads): CBG Big Data Server
[1] Sameith K, Roscito J, Hiller M. Iterative error correction of long sequencing reads maximizes accuracy and improves contig assembly. Briefings in Bioinformatics, 18(1), 1-8, 2017

PARASOL FOR LSF AND SLURM
In Bioinformatics, Genomics and many other areas, computational tasks often consist of thousands of independent jobs such as testing different parameter combinations or aligning different things against each other.
Michael wrote ParasolLSF and ParasolSlurm, a wrapper around the LSF / Slurm job queuing system that allows managing such big batches of independent jobs. Inspired by Jim Kent’s Parasol queuing system, this wrapper provides many functionalities that LSF / Slurm does not provide. The wrapper can submit such batches, monitor the progress, resubmit crashed jobs and wait until all jobs are done or some crashed repeatedly. It can stop running or pending jobs, recover the crashed jobs, give runtime averages and estimates when the batch is done. And, while LSF/Slurm just deals with individual jobs, this wrapper can manage several different batches at once.
Source code: github
